Si vous avez une question technico-scientifique, vous pouvez nous la poser et nous vous donnerons une réponse personnalisée à votre email.
Envoyez-nous votre requête
Les spécialistes en renseignements médicaux de Faes Farma ont recueilli diverses questions relatives aux interactions pharmacologiques éventuelles de la bilastine. Elles sont résumées dans le tableau 1. En l’absence d’essais cliniques directs portant sur la prise concomitante de bilastine, mais en s’appuyant sur une connaissance approfondie de son profil pharmacocinétique ainsi que sur les données scientifiques disponibles à propos d’autres médicaments, des recommandations sont émises afin de permettre une décision informée à propos du rapport risques-bénéfices de certaines associations de la bilastine à d’autres médicaments.
Tableau des interactions avec d’autres médicaments
En l’absence de données cliniques, il faut faire preuve de précaution au moment de prescrire simultanément de la bilastine (BIL) et de la DIG, mais la probabilité d’une interaction semble faible. La DIG est un glucoside cardiotonique employé pour traiter l’insuffisance cardiaque qui possède une marge thérapeutique étroite. Il s’agit d’un substrat de la glycoprotéine P (P-gp). Les médicaments qui inhibent la P-gp entravent l’élimination rénale par sécrétion tubulaire de la DIG. La BIL est aussi un substrat de la P-gp, mais n’inhibe pas son action. Ainsi, il n’existe aucun fondement scientifique qui indique la BIL puisse affecter la biodisponibilité de la DIG.
Sachant qu’il n’existe pour l’heure aucune donnée pharmacocinétique relative à l’interaction de la BIL avec des antituberculeux, le médecin doit soigneusement évaluer le rapport risques-bénéfices s’il envisage l’usage simultané de ces traitements.
On utilise typiquement différentes combinaisons de rifampicine, d’isoniazide, de pyrazinamide et d’éthambutol en traitement de première intention. Les traitements de deuxième et troisième intentions incluent les aminoglycosides, les quinolones, la rifabutine et d’autres substances. La plupart de ces médicaments induisent la P-gp ou sont éliminés par voie rénale.
On ne peut écarter la possibilité que ces médicaments diminuent l’élimination de la BIL et augmentent sa concentration plasmatique.
Afin de choisir le traitement adapté sur la base d’une évaluation du rapport risques-bénéfices, le médecin doit connaître le schéma antirétroviral concret que suit le patient. Ne pas administrer de BIL en cas de troubles rénaux et si le patient suit un traitement qui inhibe la P-gp.
Les médicaments antirétroviraux disponibles sont nombreux et peuvent se combiner de différentes manières. Leur marge thérapeutique est étroite et les interactions pharmacologiques peuvent s’avérer importantes.
Si le cytochrome P450 (CYP 450) assure le métabolisme, la BIL n’a en général aucun impact. Si on l’associe à un inhibiteur de la P-gp, on peut entraîner une augmentation de la biodisponibilité de la bilastine, ce qui n’est normalement pas pertinent d’un point de vue clinique. Toutefois, la prudence est de mise chez les patients souffrant de troubles rénaux.
Il ne devrait y avoir aucune interaction entre la BIL et les IPP.
Les IPP sont des inhibiteurs du CYP 450 et cette fonction explique un bon nombre de leurs interactions pharmacologiques. Par ailleurs, les IPP inhibent aussi la P-gp, mais cet effet ne semble pas pertinent d’un point de vue clinique.
La BIL n’est pas métabolisée et il n’est pas probable qu’elle soit affectée par la prise concomitante d’IPP.
Il ne devrait y avoir aucune interaction entre la BIL et les CO. Les femmes ayant participé aux essais cliniques devaient utiliser une méthode contraceptive efficace, ce qui incluait les CO, et aucune interaction n’a été constatée.
Les CO sont métabolisés par le CYP 450, ce qui explique un bon nombre de leurs interactions avec d’autres médicaments et les risques de grossesses non désirées. En outre, les CO inhibent le CYP 450, ce qui pourrait perturber le métabolisme d’autres médicaments.
La BIL n’inhibe ni n’induit le CYP 450. De plus, la BIL n’est pas métabolisée et, par conséquent, les CO ne peuvent perturber son élimination.
Les spécialistes en renseignements médicaux de Faes Farma ont recueilli diverses questions relatives aux interactions pharmacologiques éventuelles de la bilastine. Elles sont résumées dans le tableau 1. En l’absence d’essais cliniques directs portant sur la prise concomitante de bilastine, mais en s’appuyant sur une connaissance approfondie de son profil pharmacocinétique ainsi que sur les données scientifiques disponibles à propos d’autres médicaments, des recommandations sont émises afin de permettre une décision informée à propos du rapport risques-bénéfices de certaines associations de la bilastine à d’autres médicaments.
Grossesse
Comme l’indique la fiche technique de la bilastine, il n’existe que peu ou pas de données relatives à l’utilisation de la bilastine chez les femmes enceintes. Par mesure de précaution, il vaut mieux éviter de prendre des comprimés de Bilaxten 20 mg pendant la grossesse. Il convient de décider s’il est préférable d’interrompre le traitement ou d’y renoncer en considérant les bénéfices de l’allaitement pour l’enfant et les bénéfices du traitement pour la mère.
Les directives de l’Agence européenne des médicaments (AEM) exigent de prendre en compte toutes les connaissances disponibles à l’heure de recommander l’utilisation de médicaments pour les femmes enceintes ou pratiquant l’allaitement, ainsi que chez celles en âge de procréer. L’innocuité des antihistaminiques en cas de grossesse n’a pas été complètement établie. Ces médicaments doivent s’utiliser uniquement si, conformément à l’évaluation effectuée par le médecin, les bénéfices sont supérieurs aux risques.
Les recommandations de la FDA, actuellement en révision, concernant l’utilisation de médicaments pendant la grossesse et l’allaitement emploie les catégories d’innocuité A, B, C, D et X (tableau 2).

Système nerveux central et fonctions psychomotrices
Les antihistaminiques H1 de première génération ont un effet sédatif clairement supérieur à celui des antihistaminiques H1 de deuxième génération, ce qui a des répercussions négatives sur les fonctions psychomotrices et peut affecter la réalisation de tâches comme la conduite de véhicules ou le pilotage d’avions, etc.
L’interaction entre les antihistaminiques H1 de deuxième génération et le transporteur P-gp, qui agit au niveau de la barrière hémato-encéphalique et sert de pompe d’efflux, limite l’accès de ceux-ci au cerveau. Le résultat est une diminution des concentrations dans le cerveau, ce qui réduit au minimum les effets potentiels sur le système nerveux central, notamment la sédation.
En ce qui concerne les contrôleurs aériens (ATCS, Air Traffic Control Specialists), il existe des directives relatives à l’utilisation thérapeutique de médicaments et les recommandations concernant les antihistaminiques sont les suivantes : « antihistaminiques : les anciens antihistaminiques de type sédatif et les nouveaux qui présentent encore des effets sédatifs, comme la cétirizine, ne sont pas admis. Les nouveaux antihistaminiques non sédatifs (fexofénadine, loratadine, desloratadine) peuvent être utilisés par les ATCS après la réalisation d’un examen par le Regional Flight Surgeon qui confirme l’absence d’effets secondaires indésirables risquant d’affecter leurs capacités à effectuer leurs tâches de manière sûre ».
Quant aux pilotes, sachant que les effets de certains médicaments peuvent s’intensifier en cas d’hypoxie due à l’altitude, il est important de signaler que la Federal Aviation Administration (FAA, administration fédérale de l’aviation américaine) autorise les pilotes de ligne à prendre des antihistaminiques de deuxième génération non sédatifs tels que la loratadine, la desloratadine et la fexofénadine et déconseille l’utilisation de la cétirizine et de la lévocétirizine. L’étude des effets des médicaments chez les pilotes de ligne est possible grâce à des chambres hypoxiques qui leur permettent de tester différentes tâches et risques associés au vol à haute altitude dans un environnement sûr et contrôlé.
En ce qui concerne la bilastine, une étude récente a évalué ses effets sur la capacité à réaliser des tâches complexes propres aux pilotes de ligne dans des conditions simulant une cabine pressurisée. Les auteurs sont arrivés à la conclusion que la bilastine n’induit aucune somnolence significative et n’affecte pas les facultés psychomotrices associées au pilotage d’un avion en hypoxie induite.
Les mêmes considérations s’appliquent à l’usage de la bilastine par des personnes réalisant d’autres tâches nécessitant des compétences psychomotrices telles que la conduite automobile. L’absence d’effets de la bilastine sur la capacité de conduite en situation réelle a été démontrée lors d’un test de conduite sur route. Les sujets avaient pris des doses uniques ou répétées de bilastine, ce qui suggère qu’un dosage allant jusqu’à 40 mg ne représente pas de danger dans le trafic.
Les antihistaminiques H1 de première génération ont un effet sédatif clairement supérieur à celui des antihistaminiques H1 de deuxième génération, ce qui a des répercussions négatives sur les fonctions psychomotrices et peut affecter la réalisation de tâches comme la conduite de véhicules ou le pilotage d’avions, etc.
L’interaction entre les antihistaminiques H1 de deuxième génération et le transporteur P-gp, qui agit au niveau de la barrière hémato-encéphalique et sert de pompe d’efflux, limite l’accès de ceux-ci au cerveau. Le résultat est une diminution des concentrations dans le cerveau, ce qui réduit au minimum les effets potentiels sur le système nerveux central, notamment la sédation.
Patients atteints de troubles rénaux
De manière générale, les patients transplantés souffrant de rhinoconjonctivite allergique ou d’urticaire et prenant un immunosuppresseur comme la cyclosporine ne devraient utiliser la bilastine que si leur fonction rénale est normale ou légèrement altérée, et ce, sous étroite supervision. La fiche technique de la bilastine déconseille son utilisation chez les patients prenant de la cyclosporine et souffrant de troubles rénaux allant de modérés à graves, car ce médicament est un inhibiteur puissant de la P-gp. Pour cette raison, il pourrait augmenter la concentration plasmatique de la bilastine chez ces patients.
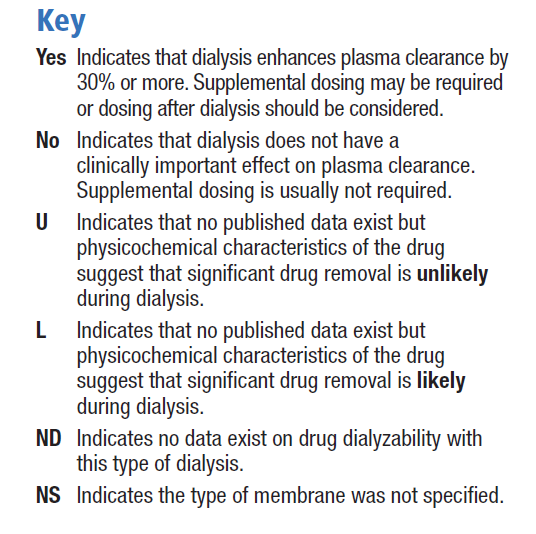
Les caractéristiques physicochimiques et propriétés pharmacocinétiques d’un médicament définissent en grande partie dans quelle mesure il sera affecté par la dialyse. En outre, d’autres aspects techniques de la dialyse peuvent aussi déterminer le degré d’élimination d’un médicament lors de ce processus. Malheureusement, peu d’études in vivo à ce sujet ont été publiées.
Par conséquent, de nombreuses directives posologiques médicamenteuses pendant l’épuration extra-rénale continue (EERC) extrapolent les résultats obtenus lors d’hémodialyses chroniques ou se basent sur des considérations théoriques. La publication Dialysis of Drugs inclut un tableau qui sert de référence en matière d’effets de la dialyse sur l’élimination des médicaments. À cet égard, il est considéré que les caractéristiques de la bilastine sont similaires à celles de la fexofénadine et que, par conséquent, la bilastine est dialysable au même titre que celle-ci, bien qu’il n’existe aucune donnée concernant des patients sous dialyse prenant de la bilastine.
Les recommandations du guide Dialisis of Drugs publiées en 2013 à propos de la fexofénadine sont les suivantes. Pour l’hémodialyse conventionnelle : NO/NS. NO signifie que la dialyse n’a aucun effet cliniquement significatif sur l’épuration plasmatique et qu’une dose supplémentaire ne devrait pas être nécesaire. NS indique que le type de membrane n’a pas été précisé. Pour l’hémodialyse à haute perméabilité : ND, ce qui signifie qu’il n’existe aucune donnée relative au caractère dialysable du médicament pour ce type de dialyse. Pour la dialyse péritonéale : U, ce qui indique qu’une élimination significative du médicament n’est pas probable étant données ses caractéristiques physicochimiques, parmi lesquelles la liaison à des protéines, la taille moléculaire ou le volume de distribution.


Certains antihistaminiques, principalement ceux de première génération, peuvent provoquer une rétention urinaire chez les patients atteints d’hypertrophie bénigne de la prostate (HBP) liée à une faible sélectivité et à l’interaction avec les récepteurs muscariniques. L’utilisation de la bilastine par un sous-groupe de patients souffrant de HBP diagnostiquée avec rétention urinaire ou dysurie n’a pas été étudiée. Cependant, étant donné sa haute sélectivité pour les récepteurs H1 de l’histamine, il apparaît raisonnable de conclure que la bilastine n’augmente pas le risque de rétention urinaire ou de dysurie.
General queries
On a constaté que les jus de fruits, notamment celui de pamplemousse, réduisent la biodisponibilité de certains médicaments, car ils inhibent le transport de captation au moyen des OATP (transporteurs d’anions organiques) 1A2. L’inhibition dure entre deux et quatre heures, ce qui indique que l’interaction est évitable en prévoyant un intervalle de temps adéquat entre la consommation de jus et la prise du médicament. Les taux plasmatiques de la bilastine sont environ 30 % inférieurs chez les sujets qui ont pris 20 mg de bilastine avec du jus de pamplemousse, comparativement à ceux qui ont pris le médicament seul. Ainsi, afin d’obtenir une efficacité maximale, il est recommandé de prendre la bilastine deux heures avant ou une heure après avoir consommé du jus de fruits ou d’autres aliments.
Étant données les propriétés pharmacocinétiques de la bilastine, il est recommandé d’attendre une période de sevrage pharmacologique de cinq jours entre la dernière dose de bilastine et la réalisation des tests d’allergie intradermiques.
Selon les résultats des tests réalisés en chambre d’exposition à Vienne, la bilastine commence à agir environ une heure après son administration.
Dans une récente étude évaluant l’inhibition de la réponse des papules et des érythèmes induite par la présence intradermique d’histamine sur des volontaires sains, il s’est avéré que la bilastine commence à agir après 30 minutes. Une heure après l’administration, on a aussi observé une diminution significative des évaluations du prurit comparativement au placébo.
De manière générale, il faut arrêter l’administration de bilastine dès la disparition des symptômes et reprendre le traitement s’ils réapparaissent. Toutefois, en cas de rhinite allergique pérenne, il est recommandé de suivre un traitement continu durant toute la période d’exposition aux allergènes. Dans les études cliniques réalisées à ce jour, la durée de traitement la plus longue était de douze mois.
Bien qu’il n’existe pas de données relatives à l’administration entérale de la bilastine, il n’y a aucune raison de penser que cette méthode présente des risques, car on a administré jusqu’à onze fois la dose recommandée sans observer aucune conséquence négative. Si la biodisponibilité est affectée par quelque raison que ce soit, l’efficacité du médicament peut diminuer quand on l’administre par voie entérale. En mai 2018, la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS, agence espagnole des médicaments et des produits pharmaceutiques) a approuvé deux nouveaux formats de la bilastine destinés aux enfants de plus de 6 ans : comprimés orodispersibles de 10 mg et solution orale de 2,5 mg/ml. Bien que ces produits conçus pour administrer une dose quotidienne de 10 mg soient destinés aux enfants âgés de 6 à 11 ans (moitié de la dose recommandée pour les adultes et adolescents), tous deux représentent une alternative envisageable pour administrer le comprimé dissous ou écrasé par sonde à des patients présentant des problèmes de déglutition.
La photosensibilisation (réactions photoallergiques) est une réaction acquise, de mécanisme immunitaire, provoquée par un médicament ou un produit chimique. Elle apparaît suite à la formation de photoproduits lorsque le médicament ou le produit chimique est exposé à des rayons ultraviolets (UVA ou UVB) ou à la lumière naturelle. Les médicaments qui ne réagissent pas aux longueurs d’onde se situant entre 290 nm et 700 nm du spectre électromagnétique ne manifestent pas de photoactivation. En d’autres termes, ils ne sont pas des photosensibilisateurs photochimiques directs.
La bilastine réagit à une longueur d’onde ultraviolette visible maximale de 253 nm. Cette valeur se situe clairement hors de l’intervalle en question et il s’avère donc inutile de procéder à une étude de photosensibilisation.
La fiche technique approuvée indique que la bilastine convient au traitement des symptômes associés à la rhinoconjonctivite allergique et à l’urticaire. Le médicament n’a pas été approuvé pour un emploi prophylactique face à ces maladies.
La dose autorisée figurant dans la fiche technique de la bilastine est de 20 mg une fois par jour. Cependant, des publications faisant l’objet d’un consensus suggèrent qu’il est possible d’administrer jusqu’à quatre fois la dose recommandée d’antihistaminiques H1 non sédatifs.
Il a été démontré que 20 mg, 40 mg et 80 mg de bilastine une fois par jour pendant sept jours se sont avérés efficaces pour abaisser le seuil de température critique chez 19 patients atteints d’urticaire au froid sur 20. En outre, pendant le programme de développement clinique de la bilastine, des chercheurs ont étudié des dosages supérieurs afin d’évaluer la pharmacocinétique, l’efficacité, la tolérance et l’innocuité de ce médicament. Ils n’ont observé aucune tendance cliniquement significative sur les ECG, dans les constantes vitales ou dans les conclusions des examens physiques. Les résultats de ces études soutiennent que la bilastine, à dose recommandée ou supérieure, même associée à du kétoconazole, est sûre d’un point de vue cardiaque. De plus, 20 mg et 40 mg de bilastine n’ont provoqué aucune altération des facultés psychomotrices ni de l’aptitude à la conduite.
Par ailleurs, tout au long du programme de développement clinique, le profil des événements indésirables de la bilastine n’a démontré aucune différence quand on le compare à celui du placébo. Si, après avoir évalué le rapport risques-bénéfices, le médecin décide de suivre les « recommandations d’augmentation du dosage » proposées pour le traitement de l’urticaire, la bilastine est une option à considérer en raison de son excellent profil d’innocuité.
Partout dans le monde, on observe des efforts croissants pour respecter l’environnement et de nouvelles questions relatives à l’impact sur celui-ci surgissent sans cesse. D’ailleurs, l’AEM exige aujourd’hui une évaluation des risques environnementaux (ERA, Environmental Risk Assessment) pour tous les nouveaux médicaments destinés aux humains. Conformément à cette exigence, on a évalué la concentration environnementale estimée de la bilastine dans les eaux de surface ainsi que son impact sur les systèmes aquatiques. Cette étude n’a soulevé aucune inquiétude. On peut donc considérer la bilastine comme respectueuse de l’environnement.


