If you have any technic-scientific question, you may ask us and we shall give you a customized answer to your email.
Send us your query
The Medical Information Specialists at Faes Farma have fielded a number of queries relating to potential drug interactions involving bilastine and these are summarised in Table 1. An area of concern relates to drugs with a narrow therapeutic index such as anticoagulants (including the novel oral anticoagulants), digoxin, and antiretroviral and antituberculosis regimens.
Whilst direct experience regarding the concomitant use of many agents with bilastine is lacking, there are data available that can help us make an informed decision regarding the riskbenefit balance of certain drug combinations. In particular, the pharmacokinetic profile of bilastine appears favourable since it undergoes negligible metabolism and is almost exclusively eliminated via renal excretion. Bilastine neither induces nor inhibits the in vitro activity of several isoenzymes from CYP 450 system. Consequently, bilastine does not interact with cytochrome metabolic pathways, and this explains the positive response given to the question, ‘Can bilastine be administered with drugs such as acenocoumarol, rivaroxaban, apixaban, clopidogrel, corticosteroids, proton pump inhibitors, and oral contraceptives?’ (Table 1). Like digoxin, bilastine is a substrate for the P-glycoprotein (P-gp) efflux transporter, and so it has the potential to interact with drugs that are eliminated via this pathway such as rivaroxaban, apixaban, digoxin, and a number of drugs that are used to treat tuberculosis (Table 1). However, since it does not inhibit the activity of the P-gp transporter, the risks with bilastine are considered to be minimal.
Interactions Table
In the absence of clinical data, caution should be exercised when coadministering BIL and DIG, but the probability of an interaction seems low.
DIG is a cardiac glycoside used in the treatment of heart failure, and it has a narrow therapeutic window. It is a substrate for P-gp, the membrane-bound transporter enzyme. Drugs which inhibit P-gp will decrease the renal tubular elimination of DIG.
BIL is also a substrate for P-gp, but it does not inhibit its action. There is no scientific rationale why BIL would affect the bioavailability of DIG.
Since pharmacokinetic data for BIL in combination with antitubercular drugs is not currently available, the doctor needs to carefully assess the overall risk-benefit if such treatment is being considered.
Classically, drugs such as rifampicin, isoniazid, pyrazinamide, and ethambutol have been used in different combinations as first-line therapy. Second-and thirdline treatments include aminoglycosides, quinolones, rifabutin and others. Many of these agents induce the efflux P-gp transporter and/or are eliminated via renal pathways.
The potential for these drugs to reduce the elimination of BIL and increase plasma levels cannot be ruled out.
To make an informed choice based on a risk-benefit assessment, the doctor needs to know the precise antiretroviral regimen the patient is receiving. BIL should not be administered when they have renal impairment and are receiving a drug which is a P-gp inhibitor.
There are a large number of available antiretroviral drugs, and they are used in different combinations. They have a narrow therapeutic window, and DIs may be important.
If metabolism is via CYP 450, then BIL will not usually have an effect. If combined with a P-gp inhibitor, there may be an increase in the bioavailability of bilastine, which is generally not clinically significant, but caution should be exercised in patients with renal impairment.
No interaction between BIL and PPIs is anticipated.
PPIs are inhibitors of CYP 450, and this explains many of their drug-drug interactions. Furthermore, PPIs also inhibit P-gp, but this effect does not appear to be clinically relevant.
BIL is not metabolised and is unlikely to be affected by concomitant PPI therapy.
No interaction between BIL and OCs is anticipated. Women included in the clinical trials’ programme were required to use an effective contraceptive method, including OCs, and no interactions were observed.
OCs are metabolised by CYP 450, and this explains many of their drug-drug interactions and the potential risk of an unwanted pregnancy. In addition, OCs inhibit CYP 450, and this might interfere with the metabolism of other drugs.
BIL does not inhibit or induce CYP 450. Furthermore, BIL is not metabolised, and so OCs cannot interfere with their elimination.
The Medical Information Specialists at Faes Farma have fielded a number of queries relating to potential drug interactions involving bilastine and these are summarised in Table 1. An area of concern relates to drugs with a narrow therapeutic index such as anticoagulants (including the novel oral anticoagulants), digoxin, and antiretroviral and antituberculosis regimens.
Whilst direct experience regarding the concomitant use of many agents with bilastine is lacking, there are data available that can help us make an informed decision regarding the riskbenefit balance of certain drug combinations. In particular, the pharmacokinetic profile of bilastine appears favourable since it undergoes negligible metabolism and is almost exclusively eliminated via renal excretion. Bilastine neither induces nor inhibits the in vitro activity of several isoenzymes from CYP 450 system. Consequently, bilastine does not interact with cytochrome metabolic pathways, and this explains the positive response given to the question, ‘Can bilastine be administered with drugs such as acenocoumarol, rivaroxaban, apixaban, clopidogrel, corticosteroids, proton pump inhibitors, and oral contraceptives?’ (Table 1). Like digoxin, bilastine is a substrate for the P-glycoprotein (P-gp) efflux transporter, and so it has the potential to interact with drugs that are eliminated via this pathway such as rivaroxaban, apixaban, digoxin, and a number of drugs that are used to treat tuberculosis (Table 1). However, since it does not inhibit the activity of the P-gp transporter, the risks with bilastine are considered to be minimal.
Pregnancy
At this point in time, we do not have sufficient information to make an informed decision about the safety of bilastine during pregnancy. Antihistamines are commonly used for the treatment of a number of common ailments in women of childbearing age with the most frequent being allergic rhinitis (AR) and urticaria. Indeed, it has been estimated that up to 20%–30% of women experience AR and 4%–7% suffer from asthma during pregnancy which, along with some dermatological complaints, make them some of the most common groups of medical conditions that complicate pregnancy. These illnesses might affect the wellbeing and quality of life of the mother since they can be highly debilitating, presenting with a range of symptoms such as nasal discharge, itching and nasal blockage or congestion in patients with AR and an array of dermatological complaints in patients with urticaria.
The safety of antihistamines has not been fully established during pregnancy and they should only be used if, in the doctor’s assessment, the benefits outweigh the risks.
Pregnancy labelling recommendations by the FDA are currently being revised. The old system used pregnancy safety categories A, B, C, D, and X (Table 2), which the FDA now consider confusing. These will be discontinued, and a new narrative-based system incorporating summaries of the risks of a drug during pregnancy and discussions of the data supporting the summaries will be required. The FDA believes that these changes in labelling will provide more meaningful information for clinicians. Guidance from the EMA (European Medicines Agency) requires that all available knowledge both clinical and non-clinical should be taken into account when making recommendations for drug usage in pregnant or lactating women, and in women of childbearing potential. This should include an integrated evaluation of non-clinical and clinical data, which includes consideration of non-clinical pharmacological and pharmacokinetic properties of the medicinal product, as well as results from non-clinical toxicity studies and of clinical experience/knowledge about compounds within the same class.

Psychomotor performance
First-generation H1-antihistamines have a much greater sedative effect than the second-generation H1-antihistamines, and this explains their negative impact on psychomotor performance. This appears to relate to the interaction between second-generation H1-antihistamines and the P-gp transporter, which is expressed in the blood-brain barrier and acts as an efflux pump. The net result is decreased concentrations of second-generation H1-antihistamines in the brain which minimises possible central nervous system effects such as sedation.
According to US Federal Aviation Administration (FAA) advice to pilots, some people are under the impression that if a drug is safe enough to be bought over the counter, without a prescription, then it must also be safe enough to pilot an aircraft whilst under its influence. Statistics show that about 80% of all major aircraft accidents, both military and civilian, usually involve human factors.
For Air Traffic Control Specialists (ATCSs), therapeutic drug guidelines are available, and the recommendations for antihistamines are ‘Antihistamines: Older, sedating type antihistamines (e.g., chlorpheniramine [chlorphenamine; Chlor- Trimeton®, Teldrin®], diphenhydramine [Benadryl®]) and the newer, but still sedating drugs like cetirizine (Zyrtec®), are not acceptable. The newer, non-sedating antihistamines (e.g., fexofenadine [Allegra®], loratadine [Claritin®], desloratadine [Clarinex®]) including decongestant combinations, are acceptable for use by working ATCSs after review by the Regional Flight Surgeon confirming the absence of adverse side effects during a brief trial of the drug. The condition must not adversely affect the ability of the ATCS to perform safely.’
In the case of pilots, and since the effects of a particular drug can be intensified with altitude, it is important that the individual is aware of the effects that any medication might have. Despite the recognised concerns with sedating first-generation antihistamines (e.g. diphenhydramine, chlorphenamine), evidence of inappropriate use was found in 4%–11% of pilot fatalities recorded in civil aviation accidents between 1990 and 2005. Moreover, the use of antihistamines, with or without other drugs and/or alcohol, was considered to be the principal cause in 13 cases, and as a concurrent factor in another 50 cases, out of 338 aviation accidents. Indeed, diphenhydramine is the most common drug found in pilots who have died in aviation accidents.
If the pilot has any concerns about a particular drug then they should consult their local Aviation Medical Examiner (AME). Importantly, the US FAA accepts the use of the secondgeneration non-sedating antihistamines such as loratadine, desloratadine and fexofenadine by airline pilots. The study of antihistamines in airline pilots is facilitated by using altitude chambers (such as the TNO altitude chamber at The Royal Netherlands Center for Man and Aviation facility) which are valuable training devices, providing aviators with the opportunity to experience many of the hazards of high-altitude flight whilst in a controlled and safe environment. Altitude chambers allow study participants to experience unpressurised flight conditions: gas expansion, rapid decompression, hypoxia, and the use of oxygen equipment.
In a study by Valk and colleagues, a single dose of desloratadine 5 mg had no detrimental effects, over a 6-hour period (Stanford Sleepiness Scale 1, 2, 3, 5 and 6 hours after treatment), on sleepiness and performance of tasks associated with flying ability under conditions of simulated cabin pressure (8000 feet [2500 metres]; hypobaric chamber 564 mmHg [75 kPa]). In contrast, patients receiving the first-generation H1-antihistamine diphenhydramine 50 mg showed significant sleepiness at each time point from 1 hour up to, and including, 5 hours.
With respect to bilastine, a recent study (BISCAT), conducted under the supervision of Pierre Valk (TNO, Soesterberg, theNetherlands), evaluated the effects of a single dose of 20 mg on flying ability in healthy male volunteers under conditions of simulated cabin pressure. This was a phase IV, single-centre, randomised, double-blind, placebo-controlled crossover study using single doses of bilastine 20 mg, hydroxyzine 50 mg (as a positive control) and placebo. Between treatment phases, individuals were subjected to a washout period of at least 1 week. Under simulated cabin altitude, subjects were evaluated using the following standard assessment tools: Vigilance and Tracking Task (VigTrack), measuring vigilance and tracking performance (analogous to in-flight tasks undertaken by fighter pilots), and the Multi-Attribute Task (MAT) battery, measuring ability to perform multiple tasks simultaneously (similar to in-flight activities that aircraft crew members have to perform), and the Stanford Sleepiness Scale, measuring subjective sleepiness. For all three assessment tools, the effect of bilastine was similar to that of placebo over the entire 6-hour study period. In contrast, hydroxyzine significantly impaired the ability of subjects to perform each of the evaluative tasks and resulted in a significantly greater increase in sleepiness scores.
As is the case for all second-generation antihistamines, it is prudent to advise patients for whom psychomotor performance is critical to undertake a trial of treatment to establish their individual response to bilastine prior to performing such tasks. It is also worth informing them that rarely some individuals may experience drowsiness which may have a detrimental effect on their psychomotor skills.
The same considerations apply to the use of bilastine in individuals performing other tasks requiring psychomotor skills such as driving a car. The ‘real-life’ effect of bilastine on driving performance was assessed in an on-the-road driving test in a double-blind, four-way crossover study in 22 healthy volunteers who randomly received the recommended dose of bilastine (20 mg) or double the recommended dose (40 mg) once-daily, hydroxyzine 50 mg (active control) or placebo for 8 consecutive days [25]. The primary efficacy variable, SDLP (standard deviation of lateral position) in the road-tracking test, was assessed on the first and eighth day of each treatment period. There were no significant differences between bilastine 20 mg or double the recommended dosage (40 mg) and placebo in terms of SDLP values on days 1 and 8. In contrast, despite a reduction in the degree of driving impairment by the eighth day of treatment, SDLP was significantly increased with hydroxyzine compared with placebo on both evaluation days, demonstrating a lack of tolerance over time to its sedative effects. Importantly, there were no effects on driving performance after single and repeated doses of bilastine, thus suggesting its safe use in traffic at doses up to 40 mg.
In a positron emission tomography (PET) study in healthy volunteers, the brain histamine-1 receptor occupancy of bilastine (after a 20 mg dose) was significantly lower than that of hydroxyzine (25 mg) in all five regions of the cerebral cortex. Bilastine was not associated with subjective sedation or objective impairment of psychomotor performance. This demonstrates the low potential for bilastine to penetrate the blood-brain barrier. Thus, bilastine has satisfied relevant pharmacological, clinical (subjective and objective) and PET criteria to be considered as a reliable non-sedating antihistamine.
First-generation H1-antihistamines have a much greater sedative effect than the second-generation H1-antihistamines, and this explains their negative impact on psychomotor performance. This appears to relate to the interaction between second-generation H1-antihistamines and the P-gp transporter, which is expressed in the blood-brain barrier and acts as an efflux pump. The net result is decreased concentrations of second-generation H1-antihistamines in the brain which minimises possible central nervous system effects such as sedation.
Patients with renal disorders
Cyclosporine, which is used in immunosuppressive regimens in kidney-transplant patients, can cause deterioration in renal function, and it has been shown to interact with drugs metabolised via the CYP P450 system. This is not a concern for bilastine which is not metabolised and does not show any interaction with CYP 450. Cyclosporine is also a potent P-gp inhibitor, and for this reason, it might increase a drug’s plasma concentration when drug elimination is P-gp mediated.
Overall, the use of bilastine in patients with allergic rhinoconjunctivitis or urticaria undergoing organ or tissue transplantation and treated with an immunosuppressant such as cyclosporine would only be recommended in individuals with normal or mildly impaired renal function and who are being closely monitored. The bilastine summary of product characteristics (SmPC) advises that the drug should not be used in patients treated with cyclosporine with moderate to severe renal impairment.
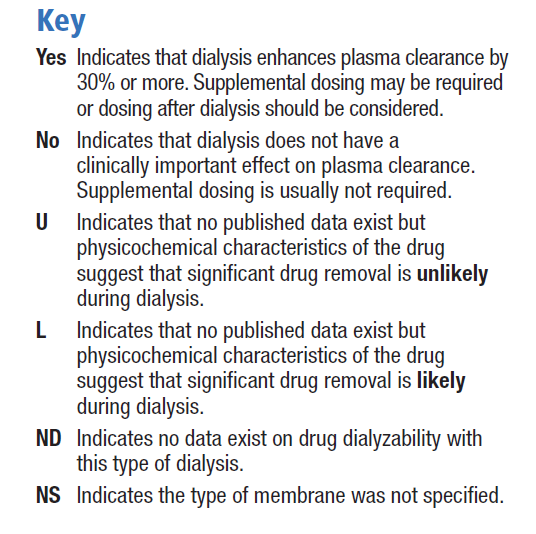
The extent to which a drug is affected by dialysis is determined primarily by its physicochemical characteristics such as molecular size and water solubility, and its pharmacokinetic properties such as absorption, plasma clearance, protein binding and volume of distribution. In addition to these properties of the drug, technical aspects of the dialysis procedure may also determine the extent to which a drug is removed by dialysis [30]. Unfortunately, few in vivo studies have been published, and the pharmacokinetic behaviour of only a small number of drugs has been investigated in dialysis patients. Therefore, many guidelines for drug dosing during continuous renal replacement therapy (CRRT) are extrapolated from experiences with chronic haemodialysis or from theoretical considerations [30]. The Dialysis of Drugs guidelines includes an accompanying table which is a reference regarding the effect of dialysis on drug clearance. The table includes antihistamines such as cetirizine, desloratadine and fexofenadine, but as bilastine is a new drug, there are no specific studies or recommendations. We consider its physicochemical characteristics to be similar to those of fexofenadine, and therefore the dialysability of bilastine might be similar to that of fexofenadine, although there are no data on dialysed patients being administered bilastine. The Dialysis of Drugs guidelines 2013 recommendations [30] for fexofenadine are conventional haemodialysis: NO/NS, NO indicates that dialysis does not have a clinically important effect on plasma clearance and supplemental dosing is not usually required / NS indicates that the type of membrane was not specified; high permeability haemodialysis: ND indicates that no data exist on drug dialysability with type of dialysis; peritoneal dialysis: U indicates significant drug removal is unlikely based on physicochemical characteristics of the drug such as protein binding, molecular size, or volume of distribution.


Some antihistamines, mainly first-generation products, may cause urinary retention in patients with benign prostatic hypertrophy (BPH). This is due to an interaction with muscarinic receptors and as a result of poor receptor selectivity. Bilastine has not been studied in a subgroup of subjects with a diagnosis of BPH with urinary retention and/or dysuria. Nevertheless, based on its receptor-selectivity profile, which was clearly established in preclinical studies, and the results from which are included in the SmPC section 5.1 (pharmacological properties) ‘without affinity for muscarinic receptors’, it seems reasonable to think that bilastine will not increase the risk of urinary retention/dysuria.
General queries
Fruit juices, especially grapefruit juice, have been shown to reduce the bioavailability of some drugs by inhibiting OAT (organic anion transporter) P1A2-mediated uptake transport. The flavanone naringin was the main causal component for the interaction, suggesting that other flavonoids in fruits and vegetables might also produce this effect. The duration of the inhibition lasted between 2 and 4 hours, indicating the interaction was avoidable, with an appropriate interval of time between juice and drug consumption. The plasma bilastine Cmax, AUC(0-t), and AUC(0-inf) values were approximately 33%, 24% and 24% lower, respectively, for subjects receiving bilastine 20 mg concomitantly with grapefruit, compared to bilastine alone. The in vitro inhibitory effects of bilastine on 12 human transporters were investigated and no clinically relevant changes were recorded. Furthermore, the transport of bilastine by multidrug resistance protein 1 (MDR1), breastcancer resistance protein (BCRP), OAT1, OAT3, and organic cation transporter2 (OCT2) was also investigated in vitro, and only MDR1 active transport of bilastine was considered potentially relevant. Finally, bilastine did not appear to be a substrate for BRCP, OCT2, OAT1 or OAT3, and it was concluded that clinically relevant drug-drug interactions resulting from inhibition of these drug transporters by bilastine would be unlikely. Thus, based on in vitro data, the only clinically relevant interaction between bilastine and membrane-transporter systems is with MDR1, and this would be avoided if bilastine were taken 2 hours before or 1 hour after fruit juices or food.
Based on the pharmacokinetic properties of bilastine, it is recommended that a 5-day washout period is allowed between the last dose of bilastine and performing a skin-prick allergy test.
Based upon findings from a Vienna Challenge Chamber Study, the onset of action of bilastine was approximately 1 hour after drug administration. This was based upon the first statistically significant (p<0.05) reduction in total nasal symptom score after drug application. In a recent study evaluating suppression of wheal-and-flare response induced by intradermal histamine in healthy volunteers, the greatest inhibition in wheal area was produced by bilastine, and it was significantly superior to desloratadine and rupatadine from 1 to 12 hours (p<0.001). Bilastine was also significantly superior to desloratadine and rupatadine for flare inhibition throughout the study, with an onset of action at 30 minutes and significantly reduced itching scores, as compared to placebo, 1 hour after administration.
Generally speaking, bilastine should be discontinued once symptoms have resolved, and it should be recommenced if symptoms reappear. However, in perennial allergic rhinitis, continued treatment is advisable during the period of allergen exposure. In clinical studies to date, the longest period of treatment has been 12 months in patients with perennial allergic rhinitis. In this open-phase study, following a controlled comparison of bilastine with cetirizine 10 mg and placebo, bilastine was well tolerated. On the basis of these findings, it seems reasonable to think that bilastine should be administered for as long as symptoms persist.
Whilst no data are available for the enteral administration of bilastine, there is no reason to think that safety would be an issue, since doses up to 11 times the recommended dose have been used without negative consequences. If bioavailability is compromised for any reason, then the drug may be less effective when administered by the enteral route.
Photosensitivity or photoallergic reactions are acquired, immunologically mediated reactions to a drug or chemical, initiated by the formation of photoproducts when that drug or chemical is exposed to UVA/UVB or visible light. Drug products that do not absorb between 290 and 700 nm of the electromagnetic spectrum will not be photoactivated, that is, they cannot be direct photochemical photosensitisers. The ultraviolet/visible radiation-absorption spectrum for bilastine is 253 nm, which is below the minimum of the range for which photosafety testing is recommended. Therefore, photosafety testing for bilastine has not been performed, as photoallergy is highly improbable.
The approved SmPC states that bilastine is indicated for the treatment of symptoms associated with allergic rhinoconjunctivitis and urticaria, and it is not approved for prophylactic management of these diseases.
The approved dosage of bilastine in the SmPC is 20 mg once daily. However, consensus reports have suggested that doses of non-sedating H1-antihistamines may be increased two- to four-fold the normal therapeutic dose in patients with urticaria. The Phase III clinical study performed to evaluate the efficacy of bilastine in the symptomatic treatment of urticaria exclusively used the approved 20 mg dosage. Bilastine 20 mg, 40 mg or 80 mg once daily for 7 days was effective in reducing critical temperature thresholds in 19 of 20 patients with cold contact urticaria (CCU). The increased efficacy of bilastine with four-fold up-dosing was without sedation and supports urticaria treatment guidelines. Additionally, during the clinical development programme for bilastine, supratherapeutic doses have been studied. In Phase I studies, doses up to about ten-fold higher (in single-dose and multiple-dose studies) were administered to assess pharmacokinetics, tolerability and safety. No clinically significant trends in ECG, vital signs or physical examination findings were observed. In another study, five-fold higher doses (100 mg), as well as 20 mg in combination with ketoconazole, were evaluated in a ‘Thorough QT/QTc’ study performed to assess the cardiac safety of the bilastine. Doses of bilastine up to four times the therapeutic dose (80 mg) were found to be well tolerated in CNS safety studies. The results of these studies support the fact that bilastine at therapeutic and supratherapeutic doses, and even when combined with ketoconazole, is safe from a cardiac point of view. Furthermore, bilastine 20 mg–40 mg did not cause psychomotor impairment or affect driving ability. In addition, throughout the clinical development programme, the adverse events profile of bilastine has not differed from that of placebo. Should the physician, after considering the risk-benefit ratio, decide to follow the proposed ‘Up-Dosing Guidelines’, bilastine should be considered as a possible candidate because of its excellent safety profile.
The world is trying to become more and more eco-friendly, and questions about environmental impact are being asked more frequently. Indeed, the EMA now requires an Environmental Risk Assessment (ERA) for all novel medicines for human use. In line with this requirement, the predicted environmental concentration of bilastine in surface water was determined, as well as its effect on aquatic systems. No environmental concerns were observed in this study, and bilastine can be considered eco-friendly.


